
Cell cycle checkpoints are control mechanisms in the eukaryotic cell cycle which ensure its proper progression. Each checkpoint serves as a potential termination point along the cell cycle, during which the conditions of the cell are assessed, with progression through the various phases of the cell cycle occurring only when favorable conditions are met. There are many checkpoints in the cell cycle,[1] but the three major ones are: the G1 checkpoint, also known as the Start or restriction checkpoint or Major Checkpoint; the G2/M checkpoint; and the metaphase-to-anaphase transition, also known as the spindle checkpoint.[2] Progression through these checkpoints is largely determined by the activation of cyclin-dependent kinases by regulatory protein subunits called cyclins, different forms of which are produced at each stage of the cell cycle to control the specific events that occur therein.[3][4]
Background
All living organisms are the products of repeated rounds of cell growth and division.[5] During this process, known as the cell cycle, a cell duplicates its contents and then divides in two. The purpose of the cell cycle is to accurately duplicate each organism's DNA and then divide the cell and its contents evenly between the two resulting cells. In eukaryotes, the cell cycle consists of four main stages: G1, during which a cell is metabolically active and continuously grows; S phase, during which DNA replication takes place; G2, during which cell growth continues and the cell synthesizes various proteins in preparation for division; and the M (mitosis) phase, during which the duplicated chromosomes (known as the sister chromatids) separate into two daughter nuclei, and the cell divides into two daughter cells, each with a full copy of DNA.[6] Compared to the eukaryotic cell cycle, the prokaryotic cell cycle (known as binary fission) is relatively simple and quick: the chromosome replicates from the origin of replication, a new membrane is assembled, and the cell wall forms a septum which divides the cell into two.[7]
As the eukaryotic cell cycle is a complex process, eukaryotes have evolved a network of regulatory proteins, known as the cell cycle control system, which monitors and dictates the progression of the cell through the cell cycle.[5] This system acts like a timer, or a clock, which sets a fixed amount of time for the cell to spend in each phase of the cell cycle, while at the same time it also responds to information received from the processes it controls. The cell cycle checkpoints play an important role in the control system by sensing defects that occur during essential processes such as DNA replication or chromosome segregation, and inducing a cell cycle arrest in response until the defects are repaired.[8] The main mechanism of action of the cell cycle checkpoints is through the regulation of the activities of a family of protein kinases known as the cyclin-dependent kinases (CDKs), which bind to different classes of regulator proteins known as cyclins, with specific cyclin-CDK complexes being formed and activated at different phases of the cell cycle. Those complexes, in turn, activate different downstream targets to promote or prevent cell cycle progression.[9]
G1 (restriction) checkpoint
The G1 checkpoint, also known as the restriction point in mammalian cells and the start point in yeast, is the point at which the cell becomes committed to entering the cell cycle. As the cell progresses through G1, depending on internal and external conditions, it can either delay G1, enter a quiescent state known as G0, or proceed past the restriction point.[5] DNA damage is the main indication for a cell to "restrict" and not enter the cell cycle. The decision to commit to a new round of cell division occurs when the cell activates cyclin-CDK-dependent transcription which promotes entry into S phase. This check point ensures the further process.[10]
During early G1, there are three transcriptional repressors, known as pocket proteins, that bind to E2F transcription factors. The E2F gene family is a group of transcription factors that target many genes that are important for control of the cell cycle, including cyclins, CDKs, checkpoint regulators, and DNA repair proteins. Misregulation of the E2F family is often found in cancer cases, providing evidence that the E2F family is essential for the tight regulation of DNA replication and division.[10] The three pocket proteins are Retinoblastoma (Rb), p107, and p130, which bind to the E2F transcription factors to prevent progression past the G1 checkpoint.
The E2F gene family contains some proteins with activator mechanisms and some proteins with repressing mechanisms. P107 and p130 act as co-repressors for E2F 4 and E2F 5, which work to repress transcription of G1-to-S promoting factors. The third pocket protein, Rb, binds to and represses E2F 1, E2F 2, and E2F 3, which are the E2F proteins with activating abilities.[10]
Positive feedback plays an essential role in regulating the progression from G1 to S phase, particularly involving the phosphorylation of Rb by a Cyclin/CDK protein complex. Rb without a phosphate, or unphosphorylated Rb, regulates G0 cell cycle exit and differentiation. During the beginning of the G1 phase, growth factors and DNA damage signal for the rise of cyclin D levels, which then binds to Cdk4 and Cdk6 to form the CyclinD:Cdk4/6 complex.[11] This complex is known to inactivate Rb by phosphorylation. However, the details of Rb phosphorylation are quite complex and specific compared to previous knowledge about the G1checkpoint. CyclinD:Cdk4/6 places only one phosphate, or monophosphorylates, Rb at one of its fourteen accessible and unique phosphorylation sites. Each of the fourteen specific mono-phosphorylated isoforms has a differential binding preference to E2F family members, which likely adds to the diversity of cellular processes within the mammalian body.[11]
E2F 4 and E2F 5 are dependent on p107 and p130 to maintain their nuclear localization. However, Cyclin D:Cdk 4/6 also phosphorylates p107 and p130, a process which releases their bind from E2F 4 and 5 (which then escape to the cytoplasm), and allowing for E2F 1–3 to bind to the DNA and initiate transcription of Cyclin E.[10] Rb proteins maintain their mono-phosphorylated state during early G1 phase, while Cyclin E is accumulating and binding to Cdk2.
CyclinE:Cdk2 plays an additional important phosphorylation role in the G1-to-S transition. Particularly, CyclinE:Cdk2 promotes a positive feedback loop which creates an “all or nothing” switch. In many genetic control networks, positive feedback ensures that cells do not slip back and forth between cell cycle phases [12] Cyclin E:Cdk2 proceeds to phosphorylate Rb at all of its phosphorylation sites, also termed “hyper-phosphorylate”, which ensures complete inactivation of Rb. The hyper phosphorylation of Rb is considered the late G1 restriction point, after which the cell cannot go backwards in the cell cycle. At this point, E2F 1-3 proteins bind to DNA and transcribe Cyclin A and Cdc 6.[11]
Cyclin-dependent kinase inhibitor 1B (CDKN1B), also known as p27, binds to and prevents the activation of CyclinE:Cdk2 by inhibition. However, as Cyclin A accumulates and binds to Cdk2, they form a complex and inhibit p27. The G1 phase cyclin-dependent kinase works together with S phase cyclin-dependent kinase targeting p27 for degradation. In turn, this allows for full activation of Cyclin A:Cdk2, a complex which phosphorylates E2F 1-3 initiating their disassociation from the DNA promoter sites. This allows E2F 6–8 to bind to the DNA and inhibit transcription.[10] The negative feedback loop used to successfully inhibit the inhibitor, p27, is another essential process used by cells to ensure mono-directional movement and no backtrack through the cell cycle.
When DNA damage occurs, or when the cell detects any defects which necessitate it to delay or halt the cell cycle in G1, arrest occurs through several mechanisms. The rapid response involves phosphorylation events that initiate with either kinase ATM (Ataxia telangiectasia mutated) or ATR (Ataxia Telangiectasia and Rad3 related), which act as sensors, depending on the type of damage. These kinases phosphorylate and activate the effector kinases Chk2 and Chk1, respectively, which in turn phosphorylate the phosphatase Cdc25A, thus marking it for ubiquitination and degradation. As Cdc25A activates the previously mentioned cyclin E-CDK2 complex by removing inhibitory phosphates from CDK2, in the absence of Cdc25A, cyclin E-CDK2 remains inactive, and the cell remains in G1.
To maintain the arrest, another response is initiated, by which Chk2 or Chk1 phosphorylate p53, a tumor suppressor, and this stabilizes p53 by preventing it from binding Mdm2, a ubiquitin ligase which inhibits p53 by targeting it for degradation. The stable p53 then acts a transcriptional activator of several target genes, including p21, an inhibitor of the G1-to-S promoting complex cyclin E-CDK2. In addition, another mechanism by which p21 is activated is through the accumulation of p16 in response to DNA damage. p16 disrupts cyclin D-CDK4 complexes, thus causing the release of p21 from the complexes, which leads to the dephosphorylation and activation of Rb, which allows Rb to bind and inhibit E2F 1–3, thus keeping the cell from transitioning to S phase.[13] Recently, some aspects of this model have been disputed.[14]
G2 checkpoint

Following DNA replication in S phase, the cell undergoes a growth phase known as G2. During this time, necessary mitotic proteins are produced and the cell is once more subjected to regulatory mechanisms to ensure proper status for entry into the proliferative Mitotic (M) phase. Multiple mechanistic checkpoints are involved in this transition from G2 to M, with a common uniting factor of cyclin-Cdk activity.
Although variations in requisite cyclin-Cdk complexes exist across organisms, the necessity of the kinase activity is conserved and typically focuses on a single pairing. In fission yeast three different forms of mitotic cyclin exist, and six in budding yeast, yet the primary cyclin utilized is cyclin B.[15] Cyclin B will serve as reference for discussion of the G2/M checkpoint transition.
Similar to S Phase, G2 experiences a DNA damage checkpoint. The cell is once more examined for sites of DNA damage or incomplete replication, and the kinases ATR and ATM are recruited to damage sites. Activation of Chk1 and Chk2 also transpire, as well as p53 activation, to induce cell cycle arrest and halt progression into mitosis. An additional component of S phase, the Pre-Replicative Complex, must be inactivated via cyclin B-Cdk1 phosphorylation.[16]
As these previous checkpoints are assessed, G2 protein accumulation serves to activate cyclin B-Cdk1 activity via multiple mechanisms. CyclinA-Cdk2 activates Cdc25, an activator of cyclin B-Cdk1, which then deactivates the cyclin B-Cdk1 inhibitor, Wee1. This results in a positive feedback loop, significantly increasing cyclin B expression and Cdk1 activation. As the cell progresses through G2 and reaches the G2/M transition, the kinase Plk1 phosphorylates Wee1, which targets Wee1 for degradation via the SCF ubiquitin ligase complex.[17] An additional function of Plk1 is to activate Cdc25 through phosphorylation. The compound effect of Wee1 degradation and Cdc25 activation is the net removal of inhibitory phosphorylation from cdc2, which activates cdc2. Plk1 is activated at the G2/M transition by the Aurora A and Bora, which accumulate during G2 and form an activation complex. The Plk1-Cdc2-cdc25 complex then initiates a positive feedback loop which serves to further activate Cdc2, and in conjunction with an increase in cyclin B levels during G2, the resulting cdc2-cyclin B complexes then activate downstream targets which promote entry into mitosis.[18] The resultant Cdk1 activity also activates expression of Mem1-Fkh, a G2/M transition gene.[19] The rapid surge in cyclin B-Cdk1 activity is necessary, as M phase initiation is an all-or-nothing event engaging in hysteresis. Hysteresis of Cdk1 activity via cyclin B drives M phase entry by establishing a minimum threshold of cyclin B concentration. This exists at a level higher than the minimum needed for the continuation of M phase after entry, acting to safeguard the all-or-nothing event. This entry concentration is further increased in the case of incomplete DNA replication, adding another regulatory mechanism at the G2/M transition point.[20] The presence of hysteresis allows for M phase entry to be highly regulated as a function of cyclin B-Cdk1 activity.
The mechanisms by which mitotic entry is prevented in response to DNA damage are similar to those in the G1/S checkpoint. DNA damage triggers the activation of the aforementioned ATM/ATR pathway, in which ATM/ATR phosphorylate and activate the Chk1/Chk2 checkpoint kinases. Chk1/2 phosphorylate cdc25 which, in addition to being inhibited, is also sequestered in the cytoplasm by the 14-3-3 proteins. 14-3-3 are upregulated by p53, which, as previously mentioned, is activated by Chk1 and ATM/ATR. p53 also transactivates p21, and both p21 and the 14-3-3 in turn inhibit cyclin B-cdc2 complexes through the phosphorylation and cytoplasmic sequestering of cdc2. In addition, the inactivation of cdc25 results in its inability to dephosphorylate and activate cdc2.[21][22] Finally, another mechanism of damage response is through the negative regulation of Plk1 by ATM/ATR, which in turn results in the stabilization of Wee1 and Myt1, which can then phosphorylate and inhibit cdc2, thus keeping the cell arrested in G2 until the damage is fixed.[23]
G2–M transition in Xenopus oocytes
At the end of G2, the cell transitions into mitosis, where the nucleus divides. The G2 to M transition is dramatic; there is an all-or-nothing effect, and the transition is irreversible. This is advantageous to the cell because entering mitosis is a critical step in the life cycle of a cell. If it does not fully commit, the cell would run into many issues with partially dividing, ultimately likely leading to the cell's death.
In frog oocytes, the signal cascade is induced when progesterone binds to a membrane bound receptor. Downstream, Mos is activated. Mos then phosphorylates MEK1, which phosphorylates MAPK. MAPK serves two roles: activating the Cyclin B-Cdk1 complex to initiate entrance into mitosis and activating Mos . The activation of Mos leads to a positive feedback loop and therefore acts as “toggle switch” to create the all-or-nothing entrance into mitosis.

This feedback loop was first found by showing that MAPK-P (phosphorylated MAPK) concentrations increased in response to increasing levels of progesterone.[24] At the single cell level, each cell either had entirely phosphorylated MAPK or no phosphorylated MAPK, confirming that it acts as a switch-like mechanism in each cell. It was additionally shown that blocking Mos protein synthesis makes the MAPK-P responses more graded, showing that Mos protein synthesis is necessary for the all-or-none character of MAPK activation.[25]
Bistability
This process can be understood using unstability. Using the graph shown to the right, the Mos synthesis rate shifts as more progesterone is added. With each curve, there are stable fixed points and unstable fixed points. At the unstable fixed points, the system will push toward either one of the stable fixed points. So, the system can either be in the “on” state or the “off” state, not in between. When the progesterone level is high enough, the Mos curve is shifted higher and ultimately intersects the degradation line at only one point, so there is only one stable “on” state, indicating the entrance into mitosis.
The irreversibility we see in the Mitosis transition point comes from having high enough levels of progesterone in the cell. At high enough levels of progesterone, the system is monostable as a result of the positive feedback loop between Mapk and Mos. The point at which the system switches from bistable to monostable is called the saddle node bifurcation.
So, we can understand the all-or-nothing, irreversible response of the mitotic transition with a mathematical model of the molecular regulators as a bistable system that depends on the existence of positive feedback. The “off-state” is annihilated by a high enough level of progesterone and once the cell gets pushed past the off-state, it is then stuck in the on-state.
Hysteresis and the Novak–Tyson model
Coming from this bi-stable model, we can understand the mitotic transition as relying on hysteresis to drive it. Hysteresis is defined as the dependence of the state of a system on its history. The Novak–Tyson model is a mathematical model of cell cycle progression that predicts that irreversible transitions entering and exiting mitosis are driven by hysteresis. The model has three basic predictions that should hold true in cycling oocyte extracts whose cell cycle progression is dependent on hysteresis:[26]
- The concentration of cyclin B necessary to enter mitosis is higher than the concentration needed to hold a mitotic extract in mitosis.
- Unreplicated DNA raises the level of cyclin necessary for Cdc2 activation and therefore entrance into mitosis.
- There is a decrease in the rate of Cdc2 activation at concentrations of cyclin B just above the activation threshold.
Sha et al. did experiments in Xenopus laevis egg extracts in 2003 to demonstrate this hysteretic nature.[27] Using cycling extracts, they observed that the activation threshold for Δcyclin B is between 32 and 42 nM whereas the inactivation threshold is between 16 and 24 nM Δcyclin B. Therefore, these experiments confirmed the bistability of this system and the importance of hysteresis in this cell cycle transition. At the intermediate cyclin B concentrations, either the interphase or mitotic state of the cell is possible.
Replication stress response
Since entering mitosis is a large and costly commitment for the cell, it is logical that systems would be in place to prevent premature entrance into this step. It has been shown that mistakes in previous steps, such as having unreplicated sections of DNA blocks progression in the cell cycle.[28] The Novak–Tyson model predicts this occurs via raising the level of cyclin B necessary for entrance into mitosis.[26]
Sha et al. investigated whether this was true in Xenopus egg extracts. They used aphidicolin (APH) to inhibit DNA polymerase and prevent DNA replication. When treated with Cyclin B in interphase, the threshold of activation increased to between 80 and 100 nM, as predicted by the Novak–Tyson model.[27] So, these experiments confirm that the stress of unreplicated DNA in the cell affect the hysteresis loop and result in a much higher cyclin B threshold to enter into mitosis.
Metaphase checkpoint
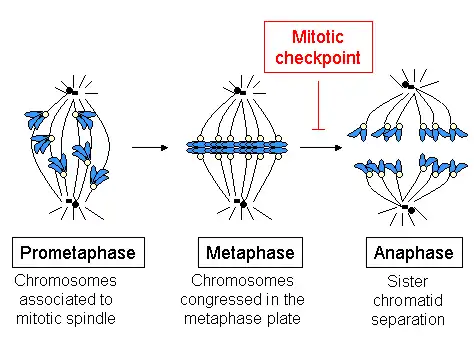
The mitotic spindle checkpoint occurs at the point in metaphase where all the chromosomes should/have aligned at the mitotic plate and be under bipolar tension. The tension created by this bipolar attachment is what is sensed, which initiates the anaphase entry. To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down securin.[29] The latter is a protein whose function is to inhibit separase, which in turn cuts the cohesins, the protein composite responsible for cohesion of sister chromatids.[30] Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation.[31] After the cell has split into its two daughter cells, the cell enters G1.
Cancer
DNA repair processes and cell cycle checkpoints have been intimately linked with cancer due to their functions regulating genome stability and cell progression, respectively. The precise molecular mechanisms that connect dysfunctions in these pathways to the onset of particular cancers are not well understood in most cases.[32] The loss of ATM has been shown to precede lymphoma development presumably due to excessive homologous recombination, leading to high genomic instability.[33] Disruption of Chk1 in mice led significant misregulation of cell cycle checkpoints, an accumulation of DNA damage, and an increased incidence of tumorigenesis.[34] Single mutant inheritance of BRCA1 or BRCA2 predisposes females toward breast and ovarian cancers.[35] BRCA1 is known to be required for S and G2/M transitions, and is involved in the cellular response to DNA damage. BRCA2 is believed to be involved in homologous recombination and regulating the S-phase checkpoint, and mutations of deficiencies in BRCA2 are strongly linked to tumorigenesis.[36]
See also
References
- ↑ Hartwell, L.; Weinert, T. (3 November 1989). "Checkpoints: controls that ensure the order of cell cycle events". Science. 246 (4930): 629–634. Bibcode:1989Sci...246..629H. doi:10.1126/science.2683079. ISSN 0036-8075. PMID 2683079.
- ↑ Morgan, David Owen (1958–2007). The cell cycle : principles of control. London: New Science Press. ISBN 978-0-19-920610-0. OCLC 70173205.
- ↑ Murray, A.; Kirschner, M. (3 November 1989). "Dominoes and clocks: the union of two views of the cell cycle". Science. 246 (4930): 614–621. Bibcode:1989Sci...246..614M. doi:10.1126/science.2683077. ISSN 0036-8075. PMID 2683077.
- ↑ Morgan, David O. (November 1997). "CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors". Annual Review of Cell and Developmental Biology. 13 (1): 261–291. doi:10.1146/annurev.cellbio.13.1.261. ISSN 1081-0706. PMID 9442875.
- 1 2 3 Alberts B, Johnson A, Lewis J, Raff M, Roberts K (2007). Molecular biology of the cell (5th ed.). New York: Garland Science. ISBN 9780815341055.
- ↑ Cooper GM (2000). The cell : a molecular approach (2nd ed.). Washington (DC): ASM Press. ISBN 978-0-87893-106-4.
- ↑ Lodish H, Baltimore D, Berk A (2000). Molecular cell biology (4th ed.). New York: Scientific American Books. ISBN 978-0-7167-3136-8.
- ↑ Malumbres M, Barbacid M (March 2009). "Cell cycle, CDKs and cancer: a changing paradigm". Nature Reviews. Cancer. 9 (3): 153–66. doi:10.1038/nrc2602. PMID 19238148. S2CID 2613411.
- ↑ Vermeulen K, Van Bockstaele DR, Berneman ZN (June 2003). "The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer". Cell Proliferation. 36 (3): 131–49. doi:10.1046/j.1365-2184.2003.00266.x. PMC 6496723. PMID 12814430.
- 1 2 3 4 5 Bertoli C, Skotheim JM, de Bruin RA (August 2013). "Control of cell cycle transcription during G1 and S phases". Nature Reviews Molecular Cell Biology. 14 (8): 518–28. doi:10.1038/nrm3629. PMC 4569015. PMID 23877564.
- 1 2 3 Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF (June 2014). "Cyclin D activates the Rb tumor suppressor by mono-phosphorylation". eLife. 3. doi:10.7554/eLife.02872. PMC 4076869. PMID 24876129.
- ↑ Skotheim JM, Di Talia S, Siggia ED, Cross FR (July 2008). "Positive feedback of G1 cyclins ensures coherent cell cycle entry". Nature. 454 (7202): 291–6. Bibcode:2008Natur.454..291S. doi:10.1038/nature07118. PMC 2606905. PMID 18633409.
- ↑ Bartek J, Lukas J (December 2001). "Mammalian G1- and S-phase checkpoints in response to DNA damage". Current Opinion in Cell Biology. 13 (6): 738–47. doi:10.1016/S0955-0674(00)00280-5. PMID 11698191.
- ↑ Bertoli C, de Bruin RA (July 2014). "Turning cell cycle entry on its head". eLife. 3: e03475. doi:10.7554/eLife.03475. PMC 4076868. PMID 24986860.
- ↑ Morgan D (2007). The Cell Cycle Principles of Control. New Science Press. pp. 92–95.
- ↑ Morgan D (2007). The Cell Cycle Principles of Control. New Science Press. pp. 228–229.
- ↑ Guardavaccaro D, Pagano M (April 2006). "Stabilizers and destabilizers controlling cell cycle oscillators". Molecular Cell. 22 (1): 1–4. doi:10.1016/j.molcel.2006.03.017. PMID 16600864.
- ↑ Seki A, Coppinger JA, Jang CY, Yates JR, Fang G (June 2008). "Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry". Science. 320 (5883): 1655–8. Bibcode:2008Sci...320.1655S. doi:10.1126/science.1157425. PMC 2834883. PMID 18566290.
- ↑ Morgan D (2007). The Cell Cycle Principles of Control. New Science Press. pp. 44–45, 90.
- ↑ Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ, Sible JC (February 2003). "Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts". Proceedings of the National Academy of Sciences of the United States of America. 100 (3): 975–80. Bibcode:2003PNAS..100..975S. doi:10.1073/pnas.0235349100. PMC 298711. PMID 12509509.
- ↑ Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W (February 2009). "Centrosome-associated regulators of the G(2)/M checkpoint as targets for cancer therapy". Molecular Cancer. 8 (1): 8. doi:10.1186/1476-4598-8-8. PMC 2657106. PMID 19216791.
- ↑ Löbrich M, Jeggo PA (November 2007). "The impact of a negligent G2/M checkpoint on genomic instability and cancer induction". Nature Reviews. Cancer. 7 (11): 861–9. doi:10.1038/nrc2248. PMID 17943134. S2CID 30207932.
- ↑ Harper JW, Elledge SJ (December 2007). "The DNA damage response: ten years after". Molecular Cell. 28 (5): 739–45. doi:10.1016/j.molcel.2007.11.015. PMID 18082599.
- ↑ Gotoh, Yukiko; Masuyama, Norihisa; Dell, Karen; Shirakabe, Kyoko; Nishida, Eisuke (October 1995). "Initiation of Xenopus Oocyte Maturation by Activation of the Mitogen-activated Protein Kinase Cascade". Journal of Biological Chemistry. 270 (43): 25898–25904. doi:10.1074/jbc.270.43.25898. ISSN 0021-9258. PMID 7592777.
- ↑ Ferrell Jr., J. E. (1998-05-08). "The Biochemical Basis of an All-or-None Cell Fate Switch in Xenopus Oocytes". Science. 280 (5365): 895–898. Bibcode:1998Sci...280..895F. doi:10.1126/science.280.5365.895. ISSN 0036-8075. PMID 9572732.
- 1 2 Novak, B.; Tyson, J.J. (1993-12-01). "Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos". Journal of Cell Science. 106 (4): 1153–1168. doi:10.1242/jcs.106.4.1153. ISSN 1477-9137. PMID 8126097.
- 1 2 Sha, W.; Moore, J.; Chen, K.; Lassaletta, A. D.; Yi, C.-S.; Tyson, J. J.; Sible, J. C. (2002-12-30). "Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts". Proceedings of the National Academy of Sciences. 100 (3): 975–980. Bibcode:2003PNAS..100..975S. doi:10.1073/pnas.0235349100. ISSN 0027-8424. PMC 298711. PMID 12509509.
- ↑ Dasso, Mary; Newport, John W. (June 1990). "Completion of DNA replication is monitored by a feedback system that controls the initiation of mitosis in vitro: Studies in Xenopus". Cell. 61 (5): 811–823. doi:10.1016/0092-8674(90)90191-g. ISSN 0092-8674. PMID 2160859. S2CID 34852886.
- ↑ Peters JM (December 1998). "SCF and APC: the Yin and Yang of cell cycle regulated proteolysis". Current Opinion in Cell Biology. 10 (6): 759–68. doi:10.1016/S0955-0674(98)80119-1. PMID 9914180.
- ↑ Ciosk R, Zachariae W, Michaelis C, Shevchenko A, Mann M, Nasmyth K (June 1998). "An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast". Cell. 93 (6): 1067–76. doi:10.1016/S0092-8674(00)81211-8. PMID 9635435. S2CID 9951929.
- ↑ Karp G (2005). Cell and Molecular Biology: Concepts and Experiments (4th ed.). Hoboken, New Jersey: John Wiley and Sons. pp. 598–9. ISBN 978-0-471-16231-5.
- ↑ Kastan MB, Bartek J (November 2004). "Cell-cycle checkpoints and cancer". Nature. 432 (7015): 316–23. Bibcode:2004Natur.432..316K. doi:10.1038/nature03097. PMID 15549093. S2CID 4415666.
- ↑ Shiloh Y, Kastan MB (2001). "ATM: genome stability, neuronal development, and cancer cross paths". Advances in Cancer Research. 83: 209–54. doi:10.1016/s0065-230x(01)83007-4. ISBN 9780120066834. PMID 11665719.
- ↑ Lam MH, Liu Q, Elledge SJ, Rosen JM (July 2004). "Chk1 is haploinsufficient for multiple functions critical to tumor suppression". Cancer Cell. 6 (1): 45–59. doi:10.1016/j.ccr.2004.06.015. PMID 15261141.
- ↑ King MC, Marks JH, Mandell JB (October 2003). "Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2". Science. 302 (5645): 643–6. Bibcode:2003Sci...302..643K. doi:10.1126/science.1088759. PMID 14576434. S2CID 33441900.
- ↑ Venkitaraman AR (January 2002). "Cancer susceptibility and the functions of BRCA1 and BRCA2". Cell. 108 (2): 171–82. doi:10.1016/s0092-8674(02)00615-3. PMID 11832208. S2CID 10397442.