I have a data frame that looks like this:
rowname Class Sec ES.2um Mean_WPBs ES.2um_ZS Mean_ES VWF_Sec name
1 Formin HAI 113.37340 147.1792 0.16078492 131.69309 112.5219 DIAPH1
2 Formin PMA 43.90661 121.9017 -0.11594028 75.37296 137.4212 FMN2
3 Septin HAI 64.32138 132.7591 -0.16218581 66.23765 150.9011 SEPTIN5
4 Septin PMA 53.15791 145.7871 -0.86969449 81.92690 140.2647 LRCH3
5 Arp2/3 PMA 68.67222 161.0516 -0.05404113 82.51804 158.2623 ARPC3
6 Arp2/3 HAI 71.00643 149.0704 -0.38119473 82.91458 130.5494 WASF3
In the "Sec" column, the row can either be "HAI" or "PMA". I am currently using gghighlight to identify a class of proteins:
plot_ll <- ggplot(df, aes(ES.2um_ZS, VWF_Sec, col = Sec,)) +
geom_point(size = 2.5) +
geom_point(aes(col=Sec)) +
geom_point() +
labs (col="Secretaogue") +
xlim(-1.25,0.7) + ylim(70,178) +
scale_colour_manual(values=c("HAI" = "blue", "PMA" = "red")) +
gghighlight(Class == "Formin", use_direct_label = FALSE, label_key = name, unhighlighted_colour = alpha("green", 0.0)) +
ggtitle("Formin Proteins Highlighted") +
theme_bw() +
theme(plot.title = element_text(hjust =0.5)) +
xlab("Mean Exit Site Z-Score (Area >2um)") + ylab("Secretion") +
geom_hline(yintercept = mean_VWF, color = "black", linetype = "dashed") +
annotate("text", -1.2, 173, label="Mean", color = "black") +
geom_vline (xintercept = mean_ES.Z, color = "black", linetype = "dashed") +
annotate("text", 0.5, 70, label = "Mean", color = "black")
Which gives something looking like this (sorry this isn't quite the same as the sample data frame above, but that data will given a similar chart for the purposes of the question):
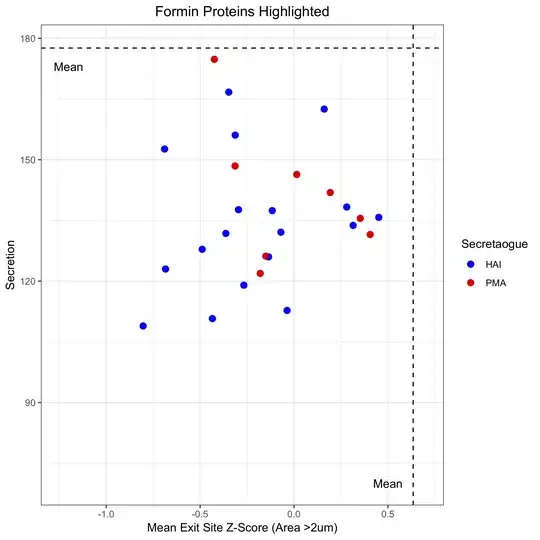
Please note that gghighlight is working, it's just the alpha value for the bleached points is set to 0 so you can't see them.
I also want to label the points with the protein names from the "name" column, so turned on use_direct_label:
plot_ll <- ggplot(df, aes(ES.2um_ZS, VWF_Sec, col = Sec,)) +
geom_point(size = 2.5) +
geom_point(aes(col=Sec)) +
geom_point() +
labs (col="Secretaogue") +
xlim(-1.25,0.7) + ylim(70,178) +
scale_colour_manual(values=c("HAI" = "blue", "PMA" = "red")) +
gghighlight(Class == "Formin", use_direct_label = TRUE, label_key = name, unhighlighted_colour = alpha("green", 0.0)) +
ggtitle("Formin Proteins Highlighted") +
theme_bw() +
theme(plot.title = element_text(hjust =0.5)) +
xlab("Mean Exit Site Z-Score (Area >2um)") +
ylab("Secretion") +
geom_hline(yintercept = mean_VWF, color = "black", linetype = "dashed") +
annotate("text", -1.2, 173, label="Mean", color = "black") +
geom_vline (xintercept = mean_ES.Z, color = "black", linetype = "dashed") +
annotate("text", 0.5, 70, label = "Mean", color = "black")
This of course gets rid of the legend. However, now there is no way to know which are "HAI" points and which are "PMA":
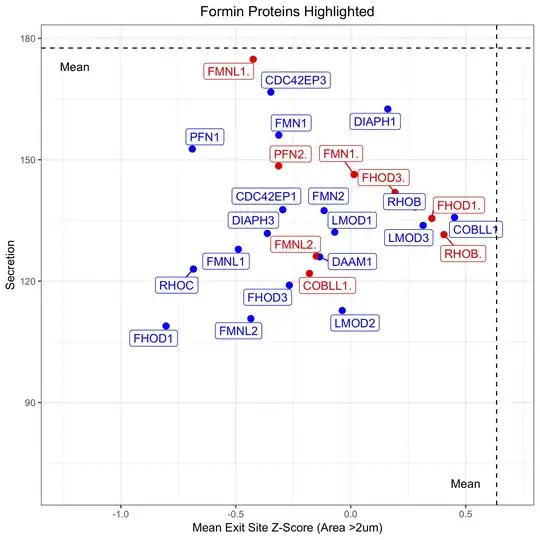
How can I retain the legend to differentiate between HAI and PMA, whilst labelling each point with the corresponding protein name?
One way might be to just manual add a legend - is this possible?
Any help would be really appreciated