The "S.h" headers file is from the "S" language (the precursor to R); replacing "S.h" with "R.h" fixes the 'cant find S.h' error, but causes other issues. Clearly this package is not being maintained :(
I've forked the repository and made a couple of changes to the source code (commits fe15cf9 and ab9fe5c). I successfully installed both the scalop and infercna packages via Bioconductor, but there are a lot of warnings when they compile. I used gcc to compile them, rather than Apple Clang, with these flags:
cat ~/.R/Makevars
LOC=/usr/local/gfortran
CC=$(LOC)/bin/gcc -fopenmp
CXX=$(LOC)/bin/g++ -fopenmp
CXX11=$(LOC)/bin/g++ -fopenmp
CFLAGS=-g -O3 -Wall -pedantic -std=gnu99 -mtune=native -pipe
CXXFLAGS=-g -O3 -Wall -pedantic -std=c++11 -mtune=native -pipe
LDFLAGS=-L$(LOC)/lib -Wl,-rpath,$(LOC)/lib,-L/usr/local/lib
CPPFLAGS=-I$(LOC)/include -I/Library/Developer/CommandLineTools/SDKs/MacOSX.sdk/usr/include -I/usr/local/include
FLIBS=-L/usr/local/gfortran/lib/gcc/x86_64-apple-darwin19/10.2.0 -L/usr/local/gfortran/lib -lgfortran -lquadmath -lm
CXX1X=/usr/local/gfortran/bin/g++
CXX98=/usr/local/gfortran/bin/g++
CXX11=/usr/local/gfortran/bin/g++
CXX14=/usr/local/gfortran/bin/g++
CXX17=/usr/local/gfortran/bin/g++
If you have problems installing the scalop package from source using Apple Clang, and you have an intel processor, my instructions for compiling R packages from source are here: https://stackoverflow.com/a/65334247/12957340
If you have an Apple silicon processor, you can try the instructions here: https://stackoverflow.com/a/68275558/12957340
This is how I installed the packages:
install.packages("BiocManager")
library(BiocManager)
BiocManager::install("Homo.sapiens")
BiocManager::install("jpmam1/scalop") # my forked copy
BiocManager::install("jlaffy/infercna")
The example from the vignette runs, but some of the functions no longer work as expected:
library(infercna)
#>
#>
#> Warning: replacing previous import 'AnnotationDbi::select' by 'dplyr::select'
#> when loading 'scalop'
#>
#> Attaching package: 'infercna'
#> The following object is masked from 'package:graphics':
#>
#> clip
set.seed(1014)
useGenome('hg19')
#> Genome has been set to hg19
retrieveGenome()
#> Retrieving: hg19
#> # A tibble: 33,575 × 8
#> symbol start_position end_position chromosome_name arm band strand
#> <chr> <dbl> <dbl> <fct> <fct> <chr> <int>
#> 1 DDX11L1 11869 14412 1 1p p36.33 1
#> 2 WASH7P 14363 29806 1 1p p36.33 -1
#> 3 MIR1302-11 29554 31109 1 1p p36.33 1
#> 4 FAM138A 34554 36081 1 1p p36.33 -1
#> 5 OR4G4P 52473 54936 1 1p p36.33 1
#> 6 OR4G11P 62948 63887 1 1p p36.33 1
#> 7 OR4F5 69091 70008 1 1p p36.33 1
#> 8 CICP27 131025 134836 1 1p p36.33 1
#> 9 RNU6-1100P 157784 157887 1 1p p36.33 -1
#> 10 CICP7 329431 332236 1 1p p36.33 -1
#> # ℹ 33,565 more rows
#> # ℹ 1 more variable: ensembl_gene_id <chr>
m = useData(mgh125)
dim(m)
#> [1] 8556 1266
range(m)
#> [1] 0.000 15.328
lengths(refCells)
#> oligodendrocytes macrophages
#> 219 707
cna = infercna(m = m, refCells = refCells, n = 5000, noise = 0.1, isLog = TRUE, verbose = FALSE)
cnaM = cna[, !colnames(cna) %in% unlist(refCells)]
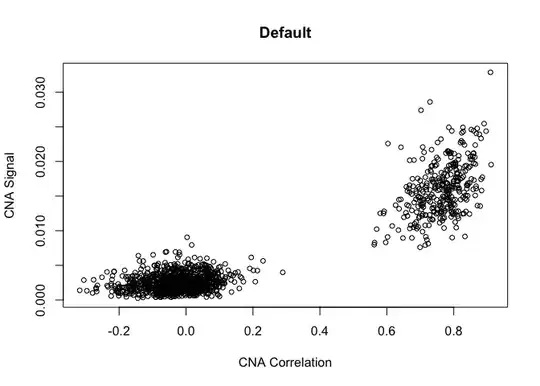
cnaScatterPlot(cna = cna,
signal.threshold = NULL,
main = 'Default')
obj = cnaPlot(cna = cna,
order.cells = TRUE,
subtitle = 'Copy-Number Aberrations in a patient with Glioblastoma')
#> Error in if (class(x) == "matrix") {: the condition has length > 1
Depending on your use-case, you'll probably need to make further changes to the source code to get your desired output. If you have further errors/questions please post them in the comments and I'll take a look at them when I have some time.
sessionInfo()
#> R version 4.3.0 (2023-04-21)
#> Platform: x86_64-apple-darwin20 (64-bit)
#> Running under: macOS Ventura 13.3.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.3-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Australia/Melbourne
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] infercna_1.0.0
#>
#> loaded via a namespace (and not attached):
#> [1] splines_4.3.0
#> [2] BiocIO_1.10.0
#> [3] bitops_1.0-7
#> [4] ggplotify_0.1.0
#> [5] filelock_1.0.2
#> [6] tibble_3.2.1
#> [7] R.oo_1.25.0
#> [8] polyclip_1.10-4
#> [9] graph_1.78.0
#> [10] reprex_2.0.2
#> [11] XML_3.99-0.14
#> [12] lifecycle_1.0.3
#> [13] rstatix_0.7.2
#> [14] edgeR_3.42.4
#> [15] Homo.sapiens_1.3.1
#> [16] lattice_0.21-8
#> [17] MASS_7.3-60
#> [18] OrganismDbi_1.42.0
#> [19] backports_1.4.1
#> [20] magrittr_2.0.3
#> [21] limma_3.56.1
#> [22] plotly_4.10.1
#> [23] rmarkdown_2.22
#> [24] yaml_2.3.7
#> [25] metapod_1.8.0
#> [26] cowplot_1.1.1
#> [27] DBI_1.1.3
#> [28] RColorBrewer_1.1-3
#> [29] abind_1.4-5
#> [30] zlibbioc_1.46.0
#> [31] Rtsne_0.16
#> [32] R.cache_0.16.0
#> [33] GenomicRanges_1.52.0
#> [34] purrr_1.0.1
#> [35] mixtools_2.0.0
#> [36] R.utils_2.12.2
#> [37] msigdbr_7.5.1
#> [38] ggraph_2.1.0
#> [39] BiocGenerics_0.46.0
#> [40] RCurl_1.98-1.12
#> [41] styler_1.10.0
#> [42] yulab.utils_0.0.6
#> [43] tweenr_2.0.2
#> [44] rappdirs_0.3.3
#> [45] GenomeInfoDbData_1.2.10
#> [46] IRanges_2.34.0
#> [47] S4Vectors_0.38.1
#> [48] enrichplot_1.20.0
#> [49] ggrepel_0.9.3
#> [50] irlba_2.3.5.1
#> [51] tidytree_0.4.2
#> [52] dqrng_0.3.0
#> [53] DelayedMatrixStats_1.22.0
#> [54] codetools_0.2-19
#> [55] DelayedArray_0.26.3
#> [56] scuttle_1.10.1
#> [57] DOSE_3.26.1
#> [58] xml2_1.3.4
#> [59] ggforce_0.4.1
#> [60] tidyselect_1.2.0
#> [61] aplot_0.1.10
#> [62] farver_2.1.1
#> [63] ScaledMatrix_1.8.1
#> [64] viridis_0.6.3
#> [65] matrixStats_0.63.0
#> [66] stats4_4.3.0
#> [67] BiocFileCache_2.8.0
#> [68] GenomicAlignments_1.36.0
#> [69] jsonlite_1.8.4
#> [70] BiocNeighbors_1.18.0
#> [71] tidygraph_1.2.3
#> [72] survival_3.5-5
#> [73] segmented_1.6-4
#> [74] tools_4.3.0
#> [75] progress_1.2.2
#> [76] treeio_1.24.1
#> [77] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
#> [78] Rcpp_1.0.10
#> [79] glue_1.6.2
#> [80] gridExtra_2.3
#> [81] xfun_0.39
#> [82] qvalue_2.32.0
#> [83] MatrixGenerics_1.12.0
#> [84] GenomeInfoDb_1.36.0
#> [85] dplyr_1.1.2
#> [86] withr_2.5.0
#> [87] BiocManager_1.30.20
#> [88] fastmap_1.1.1
#> [89] bluster_1.10.0
#> [90] fansi_1.0.4
#> [91] rsvd_1.0.5
#> [92] caTools_1.18.2
#> [93] digest_0.6.31
#> [94] R6_2.5.1
#> [95] gridGraphics_0.5-1
#> [96] colorspace_2.1-0
#> [97] GO.db_3.17.0
#> [98] biomaRt_2.56.0
#> [99] RSQLite_2.3.1
#> [100] R.methodsS3_1.8.2
#> [101] utf8_1.2.3
#> [102] tidyr_1.3.0
#> [103] generics_0.1.3
#> [104] data.table_1.14.8
#> [105] rtracklayer_1.60.0
#> [106] prettyunits_1.1.1
#> [107] graphlayouts_1.0.0
#> [108] httr_1.4.6
#> [109] htmlwidgets_1.6.2
#> [110] S4Arrays_1.0.4
#> [111] scatterpie_0.2.0
#> [112] pkgconfig_2.0.3
#> [113] gtable_0.3.3
#> [114] blob_1.2.4
#> [115] SingleCellExperiment_1.22.0
#> [116] XVector_0.40.0
#> [117] shadowtext_0.1.2
#> [118] clusterProfiler_4.8.1
#> [119] htmltools_0.5.5
#> [120] carData_3.0-5
#> [121] fgsea_1.26.0
#> [122] scalop_1.1.0
#> [123] RBGL_1.76.0
#> [124] scales_1.2.1
#> [125] Biobase_2.60.0
#> [126] png_0.1-8
#> [127] scran_1.28.1
#> [128] ggfun_0.0.9
#> [129] knitr_1.43
#> [130] rstudioapi_0.14
#> [131] reshape2_1.4.4
#> [132] rjson_0.2.21
#> [133] nlme_3.1-162
#> [134] curl_5.0.0
#> [135] org.Hs.eg.db_3.17.0
#> [136] cachem_1.0.8
#> [137] stringr_1.5.0
#> [138] parallel_4.3.0
#> [139] HDO.db_0.99.1
#> [140] AnnotationDbi_1.62.1
#> [141] restfulr_0.0.15
#> [142] pillar_1.9.0
#> [143] grid_4.3.0
#> [144] vctrs_0.6.2
#> [145] ggpubr_0.6.0
#> [146] BiocSingular_1.16.0
#> [147] car_3.1-2
#> [148] beachmat_2.16.0
#> [149] dbplyr_2.3.2
#> [150] cluster_2.1.4
#> [151] evaluate_0.21
#> [152] zeallot_0.1.0
#> [153] GenomicFeatures_1.52.0
#> [154] locfit_1.5-9.7
#> [155] cli_3.6.1
#> [156] compiler_4.3.0
#> [157] Rsamtools_2.16.0
#> [158] rlang_1.1.1
#> [159] crayon_1.5.2
#> [160] ggsignif_0.6.4
#> [161] plyr_1.8.8
#> [162] fs_1.6.2
#> [163] stringi_1.7.12
#> [164] viridisLite_0.4.2
#> [165] BiocParallel_1.34.2
#> [166] babelgene_22.9
#> [167] munsell_0.5.0
#> [168] Biostrings_2.68.1
#> [169] lazyeval_0.2.2
#> [170] GOSemSim_2.26.0
#> [171] Matrix_1.5-4.1
#> [172] patchwork_1.1.2
#> [173] hms_1.1.3
#> [174] sparseMatrixStats_1.12.0
#> [175] bit64_4.0.5
#> [176] ggplot2_3.4.2
#> [177] statmod_1.5.0
#> [178] KEGGREST_1.40.0
#> [179] SummarizedExperiment_1.30.1
#> [180] kernlab_0.9-32
#> [181] igraph_1.4.3
#> [182] broom_1.0.4
#> [183] memoise_2.0.1
#> [184] ggtree_3.8.0
#> [185] fastmatch_1.1-3
#> [186] bit_4.0.5
#> [187] downloader_0.4
#> [188] gson_0.1.0
#> [189] ape_5.7-1
Created on 2023-06-02 with reprex v2.0.2