| Baeyer-Villiger oxidation | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Named after | Adolf von Baeyer Victor Villiger | ||||||||
| Reaction type | Organic redox reaction | ||||||||
| Reaction | |||||||||
| |||||||||
| Identifiers | |||||||||
| Organic Chemistry Portal | baeyer-villiger-oxidation | ||||||||
| RSC ontology ID | RXNO:0000031 | ||||||||
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant.[1] The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.[1]

Reaction mechanism
In the first step of the reaction mechanism, the peroxyacid protonates the oxygen of the carbonyl group.[1] This makes the carbonyl group more susceptible to be attacked by the peroxyacid.[1] Next, the peroxyacid attacks the carbon of the carbonyl group forming what is known as the Criegee intermediate.[1] Through a concerted mechanism, one of the substituents on the ketone group migrates to the oxygen of the peroxide group while a carboxylic acid leaves.[1] This migration step is thought to be the rate determining step.[2][3] Finally, deprotonation of the oxocarbenium ion produces the ester.[1]
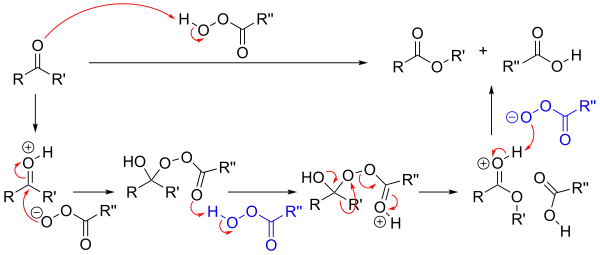
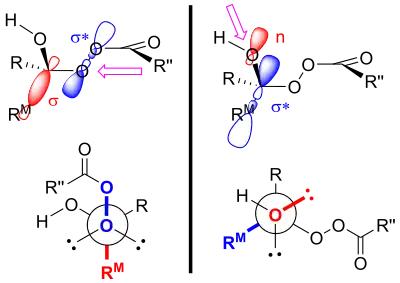
The products of the Baeyer–Villiger oxidation are believed to be controlled through both primary and secondary stereoelectronic effects.[4] The primary stereoelectronic effect in the Baeyer–Villiger oxidation refers to the necessity of the oxygen-oxygen bond in the peroxide group to be antiperiplanar to the group that migrates.[4][3] This orientation facilitates optimum overlap of the 𝛔 orbital of the migrating group to the 𝛔* orbital of the peroxide group.[1] The secondary stereoelectronic effect refers to the necessity of the lone pair on the oxygen of the hydroxyl group to be antiperiplanar to the migrating group.[4] This allows for optimum overlap of the oxygen nonbonding orbital with the 𝛔* orbital of the migrating group.[5] This migration step is also (at least in silico) assisted by two or three peroxyacid units enabling the hydroxyl proton to shuttle to its new position.[6]
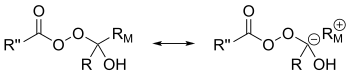
The migratory ability is ranked tertiary > secondary > aryl > primary.[7] Allylic groups are more apt to migrate than primary alkyl groups but less so than secondary alkyl groups.[5] Electron-withdrawing groups on the substituent decrease the rate of migration.[8] There are two explanations for this trend in migration ability.[9] One explanation relies on the buildup of positive charge in the transition state for breakdown of the Criegee intermediate (illustrated by the carbocation resonance structure of the Criegee intermediate).[9] Keeping this structure in mind, it makes sense that the substituent that can maintain positive charge the best would be most likely to migrate.[9] The higher the degree of substitution, the more stable a carbocation generally is.[10] Therefore, the tertiary > secondary > primary trend is observed.
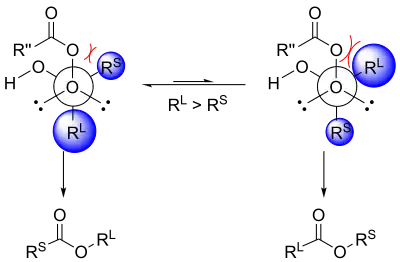
Another explanation uses stereoelectronic effects and steric arguments.[11] As mentioned, the substituent that is antiperiplanar to the peroxide group in the transition state will migrate.[4] This transition state has a gauche interaction between the peroxyacid and the non-migrating substituent.[11] If the bulkier group is placed antiperiplanar to the peroxide group, the gauche interaction between the substituent on the forming ester and the carbonyl group of the peroxyacid will be reduced.[11] Thus, it is the bulkier group that will prefer to be antiperiplanar to the peroxide group, enhancing its aptitude for migration.[11]
The migrating group in acyclic ketones, usually, is not 1° alkyl group. However, they may be persuaded to migrate in preference to the 2° or 3° groups by using CF3CO3H or BF3 + H2O2 as reagents.[12]
Historical background
In 1899, Adolf Baeyer and Victor Villiger first published a demonstration of the reaction that we now know as the Baeyer–Villiger oxidation.[13][14] They used peroxymonosulfuric acid to make the corresponding lactones from camphor, menthone, and tetrahydrocarvone.[14][15]

There were three suggested reaction mechanisms of the Baeyer–Villiger oxidation that seemed to fit with observed reaction outcomes.[16] These three reaction mechanisms can really be split into two pathways of peroxyacid attack – on either the oxygen or the carbon of the carbonyl group.[17] Attack on oxygen could lead to two possible intermediates: Baeyer and Villiger suggested a dioxirane intermediate, while Georg Wittig and Gustav Pieper suggested a peroxide with no dioxirane formation.[17] Carbon attack was suggested by Rudolf Criegee.[17] In this pathway, the peracid attacks the carbonyl carbon, producing what is now known as the Criegee intermediate.[17]

In 1953, William von Eggers Doering and Edwin Dorfman elucidated the correct pathway for the reaction mechanism of the Baeyer–Villiger oxidation by using oxygen-18-labelling of benzophenone.[16] The three different mechanisms would each lead to a different distribution of labelled products. The Criegee intermediate would lead to a product only labelled on the carbonyl oxygen.[16] The product of the Wittig and Pieper intermediate is only labeled on the alkoxy group of the ester.[16] The Baeyer and Villiger intermediate leads to a 1:1 distribution of both of the above products.[16] The outcome of the labelling experiment supported the Criegee intermediate,[16] which is now the generally accepted pathway.[1]

Stereochemistry
The migration does not change the stereochemistry of the group that transfers, i.e.: it is stereoretentive.[18][19]
Reagents
Although many different peroxyacids are used for the Baeyer–Villiger oxidation, some of the more common oxidants include meta-chloroperbenzoic acid (mCPBA) and trifluoroperacetic acid (TFPAA).[2] The general trend is that higher reactivity is correlated with lower pKa (i.e.: stronger acidity) of the corresponding carboxylic acid (or alcohol in the case of the peroxides).[5] Therefore, the reactivity trend shows TFPAA > 4-nitroperbenzoic acid > mCPBA and performic acid > peracetic acid > hydrogen peroxide > tert-butyl hydroperoxide.[5] The peroxides are much less reactive than the peroxyacids.[2] The use of hydrogen peroxide even requires a catalyst.[7][20] In addition, using organic peroxides and hydrogen peroxide tends to generate more side-reactivity due to their promiscuity.[21]
Limitations
The use of peroxyacids and peroxides when performing the Baeyer–Villiger oxidation can cause the undesirable oxidation of other functional groups.[22] Alkenes and amines are a few of the groups that can be oxidized.[22] For instance, alkenes in the substrate, particularly when electron-rich, may be oxidized to epoxides.[22][23] However, methods have been developed that will allow for the tolerance of these functional groups.[22] In 1962, G. B. Payne reported that the use of hydrogen peroxide in the presence of a selenium catalyst will produce the epoxide from alkenyl ketones, while use of peroxyacetic acid will form the ester.[24]

Modifications
Catalytic Baeyer-Villiger oxidation
The use of hydrogen peroxide as an oxidant would be advantageous, making the reaction more environmentally friendly as the sole byproduct is water.[7] Benzeneseleninic acid derivatives as catalysts have been reported to give high selectivity with hydrogen peroxide as the oxidant.[25] Another class of catalysts which show high selectivity with hydrogen peroxide as the oxidant are solid Lewis acid catalysts such as stannosilicates.[26] Among stannosilicates, particularly the zeotype Sn-beta and the amorphous Sn-MCM-41 show promising activity and close to full selectivity towards the desired product.[27][28]
Asymmetric Baeyer-Villiger oxidation
There have been attempts to use organometallic catalysts to perform enantioselective Baeyer–Villiger oxidations.[7] The first reported instance of one such oxidation of a prochiral ketone used dioxygen as the oxidant with a copper catalyst.[23] Other catalysts, including platinum and aluminum compounds, followed.[23]
Baeyer-Villiger monooxygenases

In nature, enzymes called Baeyer-Villiger monooxygenases (BVMOs) perform the oxidation analogously to the chemical reaction.[29] To facilitate this chemistry, BVMOs contain a flavin adenine dinucleotide (FAD) cofactor.[30] In the catalytic cycle (see figure on the right), the cellular redox equivalent NADPH first reduces the cofactor, which allows it subsequently to react with molecular oxygen. The resulting peroxyflavin is the catalytic entity oxygenating the substrate, and theoretical studies suggest that the reaction proceeds through the same Criegee intermediate as observed in the chemical reaction.[31] After the rearrangement step forming the ester product, a hydroxyflavin remains, which spontaneously eliminates water to form oxidized flavin, thereby closing the catalytic cycle.
BVMOs are closely related to the flavin-containing monooxygenases (FMOs),[32] enzymes that also occur in the human body, functioning within the frontline metabolic detoxification system of the liver along the cytochrome P450 monooxygenases.[33] Human FMO5 was in fact shown to be able to catalyse Baeyer-Villiger reactions, indicating that the reaction may occur in the human body as well.[34]
BVMOs have been widely studied due to their potential as biocatalysts, that is, for an application in organic synthesis.[35] Considering the environmental concerns for most of the chemical catalysts, the use of enzymes is considered a greener alternative.[29] BVMOs in particular are interesting for application because they fulfil a range of criteria typically sought for in biocatalysis: besides their ability to catalyse a synthetically useful reaction, some natural homologs were found to have a very large substrate scope (i.e. their reactivity was not restricted to a single compound, as often assumed in enzyme catalysis),[36] they can be easily produced on a large scale, and because the three-dimensional structure of many BVMOs has been determined, enzyme engineering could be applied to produce variants with improved thermostability and/or reactivity.[37][38] Another advantage of using enzymes for the reaction is their frequently observed regio- and enantioselectivity, owed to the steric control of substrate orientation during catalysis within the enzyme's active site.[29][35]
Applications
Zoapatanol
Zoapatanol is a biologically active molecule that occurs naturally in the zeopatle plant, which has been used in Mexico to make a tea that can induce menstruation and labor.[39] In 1981, Vinayak Kane and Donald Doyle reported a synthesis of zoapatanol.[40][41] They used the Baeyer–Villiger oxidation to make a lactone that served as a crucial building block that ultimately led to the synthesis of zoapatanol.[40][41]

Steroids
In 2013, Alina Świzdor reported the transformation of the steroid dehydroepiandrosterone to anticancer agent testololactone by use of a Baeyer–Villiger oxidation induced by fungus that produces Baeyer-Villiger monooxygenases.[42]

See also
References
- 1 2 3 4 5 6 7 8 9 Kürti, László; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington; San Diego; London: Elsevier Academic Press. p. 28. ISBN 978-0-12-369483-6.
- 1 2 3 Krow, Grant R. (1993). "The Baeyer-Villiger Oxidation of Ketones and Aldehydes". Organic Reactions. 43 (3): 251–798. doi:10.1002/0471264180.or043.03. ISBN 0471264180.
- 1 2 Carey, Francis A.; Sundberg, Richard J. (2007). Advanced Organic Chemistry: Part B: Reactions and Synthesis (5th ed.). New York: Springer. p. 1135. ISBN 978-0387683546.
- 1 2 3 4 Crudden, Cathleen M.; Chen, Austin C.; Calhoun, Larry A. (2000). "A Demonstration of the Primary Stereoelectronic Effect in the Baeyer-Villiger Oxidation of α-Fluorocyclohexanones". Angew. Chem. Int. Ed. 39 (16): 2851–2855. doi:10.1002/1521-3773(20000818)39:16<2851::aid-anie2851>3.0.co;2-y. PMID 11027987.
- 1 2 3 4 Myers, Andrew G. "Chemistry 115 Handouts: Oxidation" (PDF). Harvard University.
- ↑ Yamabe, Shinichi (2007). "The Role of Hydrogen Bonds in Baeyer−Villiger Reactions". The Journal of Organic Chemistry. 72 (8): 3031–3041. doi:10.1021/jo0626562. PMID 17367197.
- 1 2 3 4 ten Brink, G.-J.; Arends, W. C. E.; Sheldon, R. A. (2004). "The Baeyer-Villiger Reaction: New Developments toward Greener Procedures". Chem. Rev. 104 (9): 4105–4123. doi:10.1021/cr030011l. PMID 15352787.
- ↑ Li, Jie Jack; Corey, E. J., eds. (2007). Name Reactions of Functional Group Transformations. Hoboken, NJ: Wiley-Interscience.
- 1 2 3 Hawthorne, M. Frederick; Emmons, William D.; McCallum, K. S. (1958). "A Re-examination of the Peroxyacid Cleavage of Ketones. I. Relative Migratory Aptitudes". J. Am. Chem. Soc. 80 (23): 6393–6398. doi:10.1021/ja01556a057.
- ↑ Jones, Jr., Maitland; Fleming, Steven A. (2010). Organic Chemistry (4th ed.). Canada: W. W. Norton & Company. p. 293. ISBN 978-0-393-93149-5.
- 1 2 3 4 Evans, D. A. "Stereoelectronic Effects-2". Chemistry 206 (Fall 2006-2007).
- ↑ Sanyal, S.N. (2003). Reactions, Rearrangements and Reagents (4 ed.). p. 90. ISBN 978-81-7709-605-7.
- ↑ Baeyer, Adolf; Villiger, Victor (1899). "Einwirkung des Caro'schen Reagens auf Ketone". Ber. Dtsch. Chem. Ges. 32 (3): 3625–3633. doi:10.1002/cber.189903203151.
- 1 2 Hassall, C. H. (1957). "The Baeyer-Villiger Oxidation of Aldehydes and Ketones". Organic Reactions. 9 (3): 73–106. doi:10.1002/0471264180.or009.03. ISBN 0471264180.
- ↑ Renz, Michael; Meunier, Bernard (1999). "100 Years of Baeyer-Villiger Oxidations". Eur. J. Org. Chem. 1999 (4): 737–750. doi:10.1002/(SICI)1099-0690(199904)1999:4<737::AID-EJOC737>3.0.CO;2-B.
- 1 2 3 4 5 6 Doering, W. von E.; Dorfman, Edwin (1953). "Mechanism of the Peracid Ketone-Ester Conversion. Analysis of Organic Compounds for Oxygen-18". J. Am. Chem. Soc. 75 (22): 5595–5598. doi:10.1021/ja01118a035.
- 1 2 3 4 Doering, W. von E.; Speers, Louise (1950). "The Peracetic Acid Cleavage of Unsymmetrical Ketones". Journal of the American Chemical Society. 72 (12): 5515–5518. doi:10.1021/ja01168a041.
- ↑ Turner, Richard B. (1950). "Stereochemistry of the Peracid Oxidation of Ketones". J. Am. Chem. Soc. 72 (2): 878–882. doi:10.1021/ja01158a061.
- ↑ Gallagher, T. F.; Kritchevsky, Theodore H. (1950). "Perbenzoic Acid Oxidation of 20-Ketosteroids and the Stereochemistry of C-17". J. Am. Chem. Soc. 72 (2): 882–885. doi:10.1021/ja01158a062.
- ↑ Cavarzan, Alessandra; Scarso, Alessandro; Sgarbossa, Paolo; Michelin, Rino A.; Strukul, Giorgio (2010). "Green Catalytic Baeyer–Villiger Oxidation with Hydrogen Peroxide in Water Mediated by Pt(II) Catalysts". ChemCatChem. 2 (10): 1296–1302. doi:10.1002/cctc.201000088. S2CID 98508888.
- ↑ Schweitzer-Chaput, Bertrand; Kurtén, Theo; Klussmann, Martin (2015). "Acid-Mediated Formation of Radicals or Baeyer-Villiger Oxidation from Criegee Adducts". Angewandte Chemie International Edition. 54 (40): 11848–11851. doi:10.1002/anie.201505648. PMID 26267787.
- 1 2 3 4 Grant R. Krow (1991). Trost, Barry M.; Fleming, Ian (eds.). Comprehensive Organic Synthesis – Selectivity, Strategy and Efficiency in Modern Organic Chemistry, Volumes 1 – 9. Elsevier. pp. 671–688. ISBN 978-0-08-035930-4.
- 1 2 3 Seymour, Craig. "Page 1 The Asymmetric Baeyer-Villiger Oxidation" (PDF). scs.illinois.edu.
- ↑ Payne, G. B. (1962). "A Simplified Procedure for Epoxidation by Benzonitrile-Hydrogen Peroxide. Selective Oxidation of 2-Allylcyclohexanone". Tetrahedron. 18 (6): 763–765. doi:10.1016/S0040-4020(01)92726-7.
- ↑ ten Brink, Gerd-Jan; Vis, Jan-Martijn; Arends, Isabel W. C. E.; Sheldon, Roger A. (2001). "Selenium-Catalyzed Oxidations with Aqueous Hydrogen Peroxide. 2. Baeyer−Villiger Reactions in Homogeneous Solution". J. Org. Chem. 66 (7): 2429–2433. doi:10.1021/jo0057710. PMID 11281784.
- ↑ Ferrini, Paola; Dijkmans, Jan; Clercq, Rik De; Vyver, Stijn Van de; Dusselier, Michiel; Jacobs, Pierre A.; Sels, Bert F. (2017). "Lewis acid catalysis on single site Sn centers incorporated into silica hosts". Coordination Chemistry Reviews. 343: 220–255. doi:10.1016/j.ccr.2017.05.010.
- ↑ Corma, A; Navarro, MT; Nemeth, L; Renz, M (7 November 2001). "Sn-MCM-41—a heterogeneous selective catalyst for the Baeyer-Villiger oxidation with hydrogen peroxide". Chemical Communications (21): 2190–1. doi:10.1039/B105927K. ISSN 1364-548X. PMID 12240094.
- ↑ Renz, M; Blasco, T; Corma, A; Fornés, V; Jensen, R; Nemeth, L (18 October 2002). "Selective and shape-selective Baeyer-Villiger oxidations of aromatic aldehydes and cyclic ketones with Sn-beta zeolites and H2O2". Chemistry: A European Journal. 8 (20): 4708–17. doi:10.1002/1521-3765(20021018)8:20<4708::AID-CHEM4708>3.0.CO;2-U. ISSN 1521-3765. PMID 12561111.
- 1 2 3 Leisch, Hannes; Morley, Krista; Lau, Peter C. K. (13 July 2011). "Baeyer−Villiger Monooxygenases: More Than Just Green Chemistry". Chemical Reviews. 111 (7): 4165–4222. doi:10.1021/cr1003437. ISSN 0009-2665. PMID 21542563.
- ↑ Sheng, Dawei; Ballou, David P.; Massey, Vincent (1 September 2001). "Mechanistic Studies of Cyclohexanone Monooxygenase: Chemical Properties of Intermediates Involved in Catalysis". Biochemistry. 40 (37): 11156–11167. doi:10.1021/bi011153h. ISSN 0006-2960. PMID 11551214.
- ↑ Polyak, Iakov; Reetz, Manfred T.; Thiel, Walter (8 February 2012). "Quantum Mechanical/Molecular Mechanical Study on the Mechanism of the Enzymatic Baeyer–Villiger Reaction". Journal of the American Chemical Society. 134 (5): 2732–2741. doi:10.1021/ja2103839. ISSN 0002-7863. PMID 22239272.
- ↑ van Berkel, W. J. H.; Kamerbeek, N. M.; Fraaije, M. W. (5 August 2006). "Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts". Journal of Biotechnology. 124 (4): 670–689. doi:10.1016/j.jbiotec.2006.03.044. hdl:11370/99a1ac5c-d4a4-4612-90a3-4fe1d4d03a11. ISSN 0168-1656. PMID 16712999.
- ↑ Iyanagi, Takashi (1 January 2007). "Molecular Mechanism of Phase I and Phase II Drug‐Metabolizing Enzymes: Implications for Detoxification". International Review of Cytology. Academic Press. 260: 35–112. doi:10.1016/S0074-7696(06)60002-8. ISBN 9780123741141. PMID 17482904.
- ↑ Fiorentini, Filippo; Geier, Martina; Binda, Claudia; Winkler, Margit; Faber, Kurt; Hall, Mélanie; Mattevi, Andrea (15 April 2016). "Biocatalytic Characterization of Human FMO5: Unearthing Baeyer–Villiger Reactions in Humans". ACS Chemical Biology. 11 (4): 1039–1048. doi:10.1021/acschembio.5b01016. ISSN 1554-8929. PMID 26771671.
- 1 2 Fürst, Maximilian J. L. J.; Gran-Scheuch, Alejandro; Aalbers, Friso S.; Fraaije, Marco W. (6 December 2019). "Baeyer–Villiger Monooxygenases: Tunable Oxidative Biocatalysts". ACS Catalysis. 9 (12): 11207–11241. doi:10.1021/acscatal.9b03396.
- ↑ Fürst, Maximilian J. L. J.; Romero, Elvira; Gómez Castellanos, J. Rúben; Fraaije, Marco W.; Mattevi, Andrea (7 December 2018). "Side-Chain Pruning Has Limited Impact on Substrate Preference in a Promiscuous Enzyme". ACS Catalysis. 8 (12): 11648–11656. doi:10.1021/acscatal.8b03793. PMC 6345240. PMID 30687578.
- ↑ Fürst, Maximilian J. L. J.; Boonstra, Marjon; Bandstra, Selle; Fraaije, Marco W. (2019). "Stabilization of cyclohexanone monooxygenase by computational and experimental library design". Biotechnology and Bioengineering. 116 (9): 2167–2177. doi:10.1002/bit.27022. ISSN 1097-0290. PMC 6836875. PMID 31124128.
- ↑ Li, Guangyue; Garcia-Borràs, Marc; Fürst, Maximilian J. L. J.; Ilie, Adriana; Fraaije, Marco W.; Houk, K. N.; Reetz, Manfred T. (22 August 2018). "Overriding Traditional Electronic Effects in Biocatalytic Baeyer–Villiger Reactions by Directed Evolution". Journal of the American Chemical Society. 140 (33): 10464–10472. doi:10.1021/jacs.8b04742. ISSN 0002-7863. PMC 6314816. PMID 30044629.
- ↑ Levine, Seymour D.; Adams, Richard E.; Chen, Robert; Cotter, Mary Lou; Hirsch, Allen F.; Kane, Vinayak V.; Kanojia, Ramesh M.; Shaw, Charles; Wachter, Michael P.; Chin, Eva; Huettemann, Richard; Ostrowski, Paul (1979). "Zoapatanol and Montanol, Novel Oxepane Diterpenoids, from the Mexican Plant Zoapatle (Montanoa tomentosa)". J. Am. Chem. Soc. 101 (12): 3405–3407. doi:10.1021/ja00506a057.
- 1 2 Kane, Vinayak V.; Doyle, Donald L. (1981). "Total Synthesis of (±) Zoapatanol: A Stereospecific Synthesis of a Key Intermediate". Tetrahedron Lett. 22 (32): 3027–3030. doi:10.1016/S0040-4039(01)81818-9.
- 1 2 Kane, Vinayak V.; Doyle, Donald L. (1981). "Total Synthesis of (±) Zoapatanol". Tetrahedron Lett. 22 (32): 3031–3034. doi:10.1016/S0040-4039(01)81819-0.
- ↑ Świzdor, Alina (2013). "Baeyer-Villiger Oxidation of Some C19 Steroids by Penicillium lanosocoeruleum". Molecules. 18 (11): 13812–13822. doi:10.3390/molecules181113812. PMC 6270215. PMID 24213656.
{kind=link}