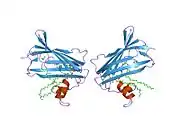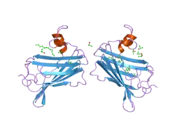




GM2 ganglioside activator also known as GM2A is a protein which in humans is encoded by the GM2A gene.[5][6]
Function
The protein encoded by this gene is a small glycolipid transport protein which acts as a substrate specific co-factor for the lysosomal enzyme β-hexosaminidase A. β-hexosaminidase A, together with GM2 ganglioside activator, catalyzes the degradation of the ganglioside GM2, and other molecules containing terminal N-acetyl hexosamines.
GM2A is a lipid transfer protein that stimulates the enzymatic processing of gangliosides, and also T-cell activation through lipid presentation. This protein binds molecules of ganglioside GM2, extracts them from membranes, and presents them to beta-hexosaminidase A for cleavage of N-acetyl-D-galactosamine and conversion to GM3.
It was identified as a member of ML domain family of proteins involved in innate immunity and lipid metabolism in the SMART database. .
Regulation
In melanocytic cells GM2A gene expression may be regulated by MITF.[7]
Clinical significance
Mutations in this gene, inherited in an autosomal recessive pattern, result in GM2-gangliosidosis, AB variant, a rare GM2 gangliosidosis that has symptoms and pathology identical with Tay–Sachs disease and Sandhoff disease.[8]
GM2A mutations are rarely reported, and the cases that are observed often occur with consanguineous parents or in genetically isolated populations.[9]
Because AB variant is so rarely diagnosed, even in infants, it is likely that most mutations of GM2A are fatal in the fetus in homozygotes and genetic compounds, and thus are never observed clinically.
See also
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000196743 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000000594 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ Li SC, Nakamura T, Ogamo A, Li YT (November 1979). "Evidence for the presence of two separate protein activators for the enzymic hydrolysis of GM1 and GM2 gangliosides". J. Biol. Chem. 254 (21): 10592–5. doi:10.1016/S0021-9258(19)86559-6. PMID 115863.
- ↑ Klima H, Tanaka A, Schnabel D, Nakano T, Schröder M, Suzuki K, Sandhoff K (September 1991). "Characterization of full-length cDNAs and the gene coding for the human GM2 activator protein". FEBS Lett. 289 (2): 260–4. doi:10.1016/0014-5793(91)81084-L. PMID 1915857. S2CID 40408626.
- ↑ Hoek KS, Schlegel NC, Eichhoff OM, et al. (2008). "Novel MITF targets identified using a two-step DNA microarray strategy". Pigment Cell Melanoma Res. 21 (6): 665–76. doi:10.1111/j.1755-148X.2008.00505.x. PMID 19067971. S2CID 24698373.
- ↑ Mahuran DJ (1999-10-08). "Biochemical consequences of mutations causing the GM2 gangliosidoses". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1455 (2–3): 105–138. doi:10.1016/S0925-4439(99)00074-5. PMID 10571007.
- ↑ "Online Mendelian Inheritance in Man". United States National Institute of Health. Retrieved 2009-04-21.
Further reading
- Li SC, Nakamura T, Ogamo A, Li YT (1980). "Evidence for the presence of two separate protein activators for the enzymic hydrolysis of GM1 and GM2 gangliosides". J. Biol. Chem. 254 (21): 10592–5. doi:10.1016/S0021-9258(19)86559-6. PMID 115863.
- Xie B, Kennedy JL, McInnes B, et al. (1992). "Identification of a processed pseudogene related to the functional gene encoding the GM2 activator protein: localization of the pseudogene to human chromosome 3 and the functional gene to human chromosome 5". Genomics. 14 (3): 796–8. doi:10.1016/S0888-7543(05)80190-9. PMID 1427911.
- Nagarajan S, Chen HC, Li SC, et al. (1992). "Evidence for two cDNA clones encoding human GM2-activator protein". Biochem. J. 282 (3): 807–13. doi:10.1042/bj2820807. PMC 1130859. PMID 1554364.
- Klima H, Tanaka A, Schnabel D, et al. (1991). "Characterization of full-length cDNAs and the gene coding for the human GM2 activator protein". FEBS Lett. 289 (2): 260–4. doi:10.1016/0014-5793(91)81084-L. PMID 1915857. S2CID 40408626.
- Schröder M, Schnabel D, Suzuki K, Sandhoff K (1991). "A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB". FEBS Lett. 290 (1–2): 1–3. doi:10.1016/0014-5793(91)81211-P. PMID 1915858. S2CID 26669859.
- Xie B, McInnes B, Neote K, et al. (1991). "Isolation and expression of a full-length cDNA encoding the human GM2 activator protein". Biochem. Biophys. Res. Commun. 177 (3): 1217–23. doi:10.1016/0006-291X(91)90671-S. PMID 2059210.
- Fürst W, Schubert J, Machleidt W, et al. (1990). "The complete amino-acid sequences of human ganglioside GM2 activator protein and cerebroside sulfate activator protein". Eur. J. Biochem. 192 (3): 709–14. doi:10.1111/j.1432-1033.1990.tb19280.x. PMID 2209618.
- Schröder M, Klima H, Nakano T, et al. (1989). "Isolation of a cDNA encoding the human GM2 activator protein". FEBS Lett. 251 (1–2): 197–200. doi:10.1016/0014-5793(89)81454-1. PMID 2753159. S2CID 22615177.
- Burg J, Banerjee A, Sandhoff K (1986). "Molecular forms of GM2-activator protein. A study on its biosynthesis in human skin fibroblasts". Biol. Chem. Hoppe-Seyler. 366 (9): 887–91. doi:10.1515/bchm3.1985.366.2.887. PMID 3935131.
- Banerjee A, Burg J, Conzelmann E, et al. (1984). "Enzyme-linked immunosorbent assay for the ganglioside GM2-activator protein. Screening of normal human tissues and body fluids, of tissues of GM2 gangliosidosis, and for its subcellular localization". Hoppe-Seyler's Z. Physiol. Chem. 365 (3): 347–56. doi:10.1515/bchm2.1984.365.1.347. PMID 6724528.
- Hirabayashi Y, Li YT, Li SC (1983). "The protein activator specific for the enzymic hydrolysis of GM2 ganglioside in normal human brain and brains of three types of GM2 gangliosidosis". J. Neurochem. 40 (1): 168–75. doi:10.1111/j.1471-4159.1983.tb12667.x. PMID 6848657. S2CID 33699881.
- Schröder M, Schnabel D, Hurwitz R, et al. (1994). "Molecular genetics of GM2-gangliosidosis AB variant: a novel mutation and expression in BHK cells". Hum. Genet. 92 (5): 437–40. doi:10.1007/BF00216446. PMID 8244332. S2CID 21839371.
- Heng HH, Xie B, Shi XM, et al. (1994). "Refined mapping of the GM2 activator protein (GM2A) locus to 5q31.3-q33.1, distal to the spinal muscular atrophy locus". Genomics. 18 (2): 429–31. doi:10.1006/geno.1993.1491. PMID 8288250.
- Klima H, Klein A, van Echten G, et al. (1993). "Over-expression of a functionally active human GM2-activator protein in Escherichia coli". Biochem. J. 292 (2): 571–6. doi:10.1042/bj2920571. PMC 1134248. PMID 8503891.
- Schepers U, Glombitza G, Lemm T, et al. (1996). "Molecular analysis of a GM2-activator deficiency in two patients with GM2-gangliosidosis AB variant". Am. J. Hum. Genet. 59 (5): 1048–56. PMC 1914821. PMID 8900233.
- Rigat B, Wang W, Leung A, Mahuran DJ (1997). "Two mechanisms for the recapture of extracellular GM2 activator protein: evidence for a major secretory form of the protein". Biochemistry. 36 (27): 8325–31. doi:10.1021/bi970571c. PMID 9204879.
- Yadao F, Hechtman P, Kaplan F (1997). "Formation of a ternary complex between GM2 activator protein, GM2 ganglioside and hexosaminidase A". Biochim. Biophys. Acta. 1340 (1): 45–52. doi:10.1016/S0167-4838(97)00027-7. PMID 9217013.
- Schütte CG, Lemm T, Glombitza GJ, Sandhoff K (1998). "Complete localization of disulfide bonds in GM2 activator protein". Protein Sci. 7 (4): 1039–45. doi:10.1002/pro.5560070421. PMC 2143992. PMID 9568910.
- Chen B, Rigat B, Curry C, Mahuran DJ (1999). "Structure of the GM2A gene: identification of an exon 2 nonsense mutation and a naturally occurring transcript with an in-frame deletion of exon 2". Am. J. Hum. Genet. 65 (1): 77–87. doi:10.1086/302463. PMC 1378077. PMID 10364519.
- Jinnai H, Nakamura S (2000). "Characterization of phospholipase D activation by GM2 activator in a cell-free system". Kobe Journal of Medical Sciences. 45 (3–4): 181–90. PMID 10752311.
PDB gallery | |
|---|---|
|