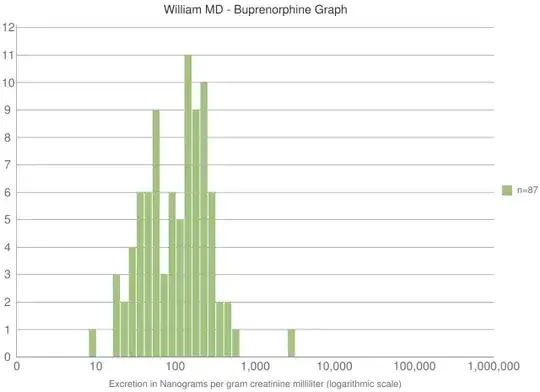
I have the following heatmap with the dendrogram like this.
The complete data are here.
The problem is that the dendrogram on the left is squished. How can I unsquish (expand) it without altering the column size of the heatmap?
It is generated with this code:
#!/usr/bin/Rscript
library(gplots);
library(RColorBrewer);
plot_hclust <- function(inputfile,clust.height,type.order=c(),row.margins=70) {
# Read data
dat.bcd <- read.table(inputfile,na.strings=NA, sep="\t",header=TRUE);
rownames(dat.bcd) <- do.call(paste,c(dat.bcd[c("Probes","Gene.symbol")],sep=" "))
dat.bcd <- dat.bcd[,!names(dat.bcd) %in% c("Probes","Gene.symbol")]
dat.bcd <- dat.bcd
# Clustering and distance function
hclustfunc <- function(x) hclust(x, method="complete")
distfunc <- function(x) dist(x,method="maximum")
# Select based on FC, as long as any of them >= anylim
anylim <- 2.0
dat.bcd <- dat.bcd[ apply(dat.bcd, 1,function(x) any (x >= anylim)), ]
# Clustering functions
height <- clust.height;
# Define output file name
heatout <- paste("tmp.pafc.heat.",anylim,".h",height,".pdf",sep="");
# Compute distance and clusteirn function
d.bcd <- distfunc(dat.bcd)
fit.bcd <- hclustfunc(d.bcd)
# Cluster by height
#cutree and rect.huclust has to be used in tandem
clusters <- cutree(fit.bcd, h=height)
nofclust.height <- length(unique(as.vector(clusters)));
myorder <- colnames(dat.bcd);
if (length(type.order)>0) {
myorder <- type.order
}
# Define colors
#hmcols <- rev(brewer.pal(11,"Spectral"));
hmcols <- rev(redgreen(2750));
selcol <- colorRampPalette(brewer.pal(12,"Set3"))
selcol2 <- colorRampPalette(brewer.pal(9,"Set1"))
sdcol= selcol(5);
clustcol.height = selcol2(nofclust.height);
# Plot heatmap
pdf(file=heatout,width=20,height=50); # for FC.lim >=2
heatmap.2(as.matrix(dat.bcd[,myorder]),Colv=FALSE,density.info="none",lhei=c(0.1,4),dendrogram="row",scale="row",RowSideColors=clustcol.height[clusters],col=hmcols,trace="none", margin=c(30,row.margins), hclust=hclustfunc,distfun=distfunc,lwid=c(1.5,2.0),keysize=0.3);
dev.off();
}
#--------------------------------------------------
# ENd of functions
#--------------------------------------------------
plot_hclust("http://pastebin.com/raw.php?i=ZaGkPTGm",clust.height=3,row.margins=70);